Bases moleculares de la enfermedad de Alzheimer
Escrito por:
Dr. Ramón Garduño Juárez
Visto: 8873 veces
Bases moleculares de la enfermedad de Alzheimer
Subramanian Boopathi y Ramón Garduño-Juárez
Subramanian Boopathi obtuvo el Ph.D. en Física por la Bharathiar Universidad, Tamilnadu, India, en 2016. Ha tenido cinco años de experiencia posdoctoral en investigación de la enfermedad de Alzheimer en Corea del Sur, Polonia, Chile y México. Su principal interés es comprender el mecanismo de agregación de los péptidos de la enfermedad de Alzheimer mediante el empleo de simulaciones computacionales. Ahora, es investigador postdoctoral de la DGAPA en la Universidad Nacional Autónoma de México.
Ramón Garduño Juárez es investigador titular de Biofísica en el Instituto de Ciencias Físicas de la Universidad Nacional Autónoma de México. Es Ingeniero Bioquímico con un Ph. D. en Biofísica de la Universidad Estatal de Nueva York en Buffalo. Fue becario posdoctoral en los Laboratorios de Investigación Ames de la NASA. Sus líneas de investigación incluyen simulaciones de dinámica molecular sobre péptidos antimicrobianos. Es miembro de la Academia Mexicana de Ciencias y de la Academia de Ciencias de Morelos.
Esta publicación fue revisada por el comité editorial de la Academia de Ciencias de Morelos.
Descripción General
La enfermedad de Alzheimer (EA) es un trastorno neurológico progresivo que hace que las neuronas cerebrales mueran y el cerebro se encoja (atrofia). La EA es la causa más común de demencia en los ancianos. Se manifiesta por un deterioro continuo en el razonamiento, el comportamiento y las habilidades sociales que afecta la capacidad de una persona para vivir de forma independiente.
Las neuronas son los componentes básicos del sistema nervioso, incluidos el cerebro y la médula espinal. Las neuronas casi no se reproducen y pierden la capacidad de hacerlo cuando están dañadas. Una combinación de factores genéticos y ambientales juega un papel importante su destrucción, así como en el desarrollo de enfermedades neurodegenerativas, que son una amenaza importante para la salud en el siglo XXI. Dado que no se conocen bien la etiología, es decir las causas, de las enfermedades neurodegenerativas es difícil identificar quién será afectado. En el mundo existen varias amenazas para la salud mental, como la enfermedad de Alzheimer, la enfermedad de Parkinson, la enfermedad priónica, la enfermedad de las neuronas motoras, la enfermedad de Huntington, la ataxia espinocerebelosa, la atrofia muscular espinal y la esclerosis lateral amiotrófica.
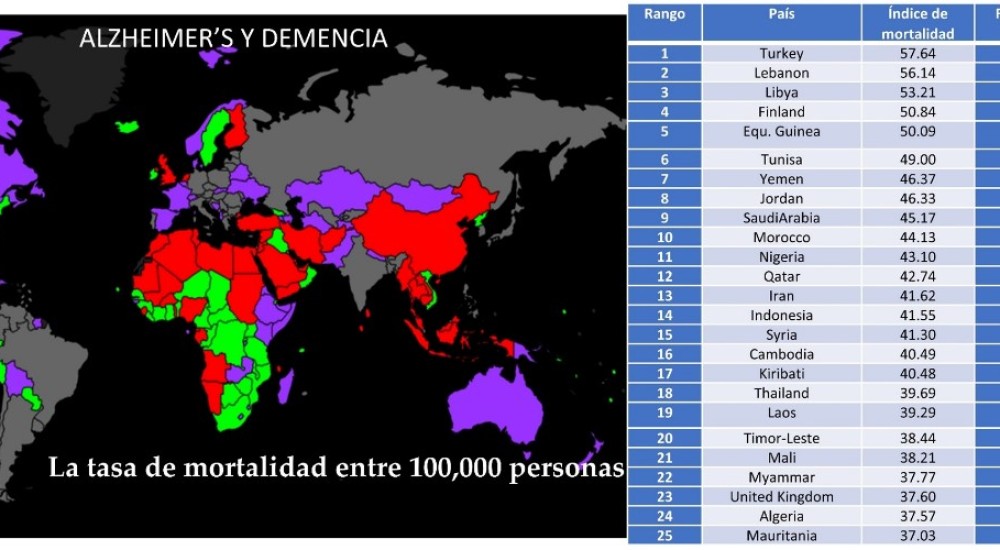
En México, la EA es la demencia más prevalente que afecta aproximadamente a 28 de cada 1000 adultos mayores cada año, y se estima que 3.5 millones la padecerán para 2050 a menos que se disponga de un tratamiento efectivo (Figura 1). Si no se conoce la etiología, no existen tratamientos específicos y su detección puede ser tardía. Hay muchas desventajas para obtener tratamiento y atención médica para esta enfermedad en México, ya que solo hay ocho casas de cuidado disponibles para 350,000 personas que han sido diagnosticadas con EA. En particular, se diagnostica un nuevo caso de demencia en todo el mundo cada tres segundos. Los principales síntomas de los pacientes con EA son la dificultad para reconocer a los miembros de la familia, leer, escribir, hablar habitualmente y el trastorno del sueño, que incluye actividades regulares perturbadoras, como cepillarse los dientes y peinarse; por lo tanto, requieren un cuidado personal completo
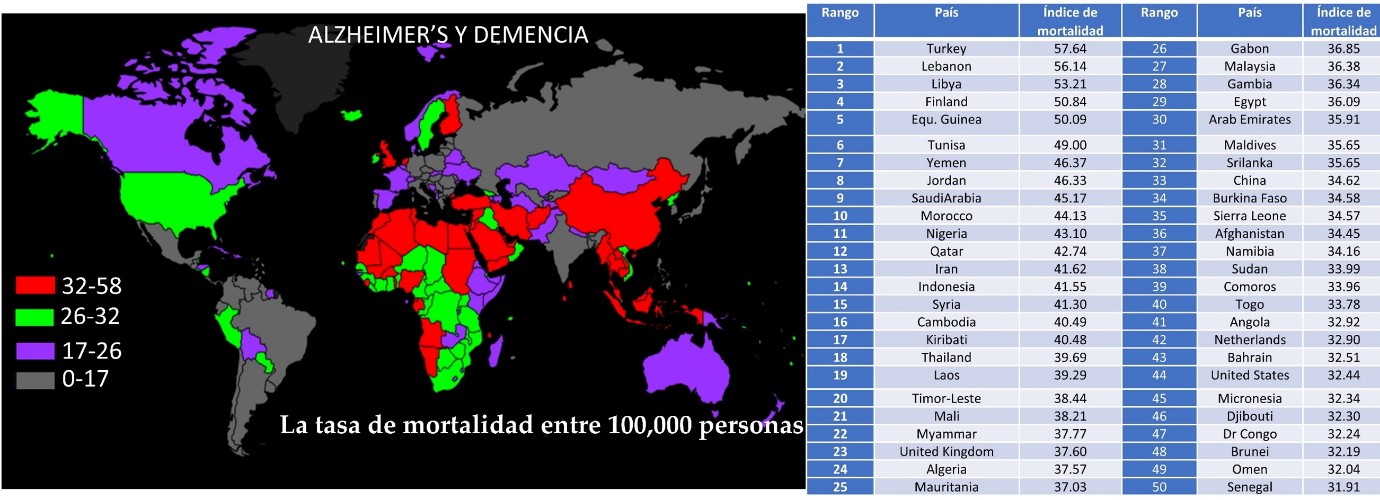
Figura 1: La Organización Mundial de la Salud (2018) informó la tasa de muerte en el país que se muestra en el mapa; La tabla tabuló los 50 países principales clasificados por tasa de mortalidad entre 100.000 personas (Adaptada de https://www.worldlifeexpectancy.com/cause-of-death/alzheimers-dementia/by-country/)
El aumento de la edad es el factor de riesgo más importante para la EA porque cada año se registran cuatro nuevos casos entre 1000 personas de 65 a 74 años; 32 entre 1000 personas de 75 a 84 años; y 76 entre 1000 personas de 85 años o más. Se ha observado que en humanos el proceso degenerativo de la EA dura de diez a veinte años hasta la muerte, y en ocasiones puede ser mucho más rápido. La EA se caracteriza por una degeneración gradual de las neuronas en el hipocampo y por una muerte lenta de éstas.
¿Qué pasa con los péptidos?
Los péptidos (ver inserción) juegan un papel muy importante en la EA. La masa total del péptido Aβ es de ~7 mg en cerebros postmortem de pacientes con EA, comparado con ~2 mg en el cerebro sano, Los ~5 mg en exceso de Aβ está ligado a las causas de la EA. Si consideramos que se depositan ~30 nanogramos por hora, se llega a los 5 mg en aproximadamente 20 años.
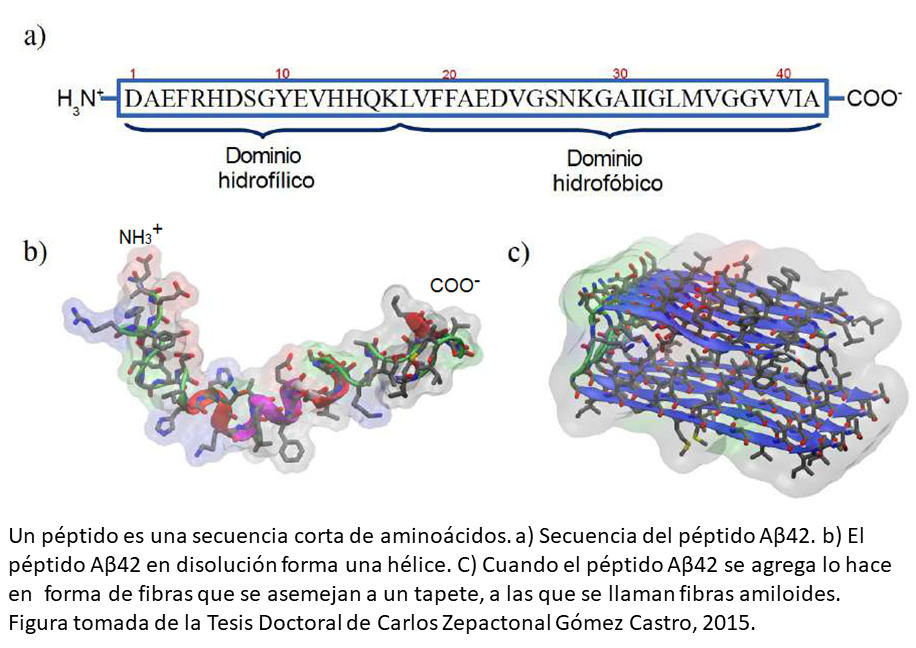
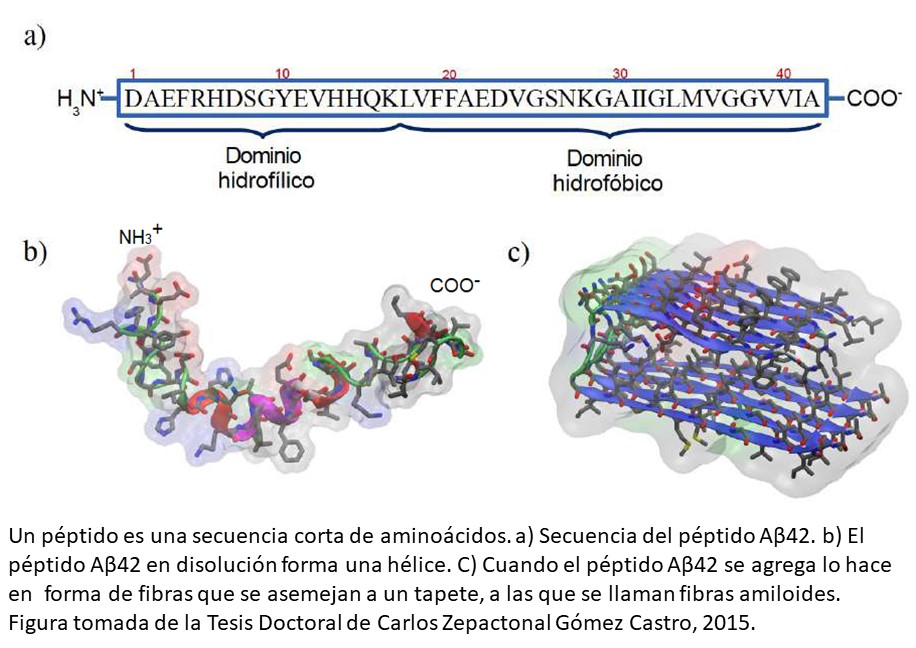
Estos depósitos ocurren debido a la disminución de la actividad de las enzimas que degradan al péptido amiloide (α-secretasa). Así, la edad avanzada favorece la formación de placas, y por lo tanto, los adultos mayores tienen un mayor riesgo de desarrollar la EA.
Las placas amiloides son agregados de péptidos amiloide β (Aβ) dentro del tejido neuronal. Los ovillos neurofibrilares están compuestos de proteína tau de fosforilación fuera de las neuronas. Tanto las placas como los ovillos impiden el funcionamiento del cerebro al romper la conexión entre las neuronas, lo que provoca un grave problema de memoria.
Existen dos formas de la EA, una es conocida como de inicio temprano o familiar y la otra de inicio tardío o esporádica. La forma familiar se caracteriza por ser hereditaria y de inicio temprano (antes 65 años). En contraste, la forma esporádica se presenta generalmente después de los 65 años. Más del 95% de los casos son esporádicos, mientras que los casos familiares son raros (< 5%) comienzan antes y están relacionados con mutaciones dominantes heredadas en la proteína precursora del amiloide responsable de generar proteínas amiloideas neurotóxicas.
¿Qué son los péptidos amiloideos neurotóxicos?
La neurotoxicidad se refiere a las neuronas que se lesionan y mueren en el hipocampo por la agregación de los péptidos β-amiloideos (Aβ), ya que el hipocampo es el principal responsable del aprendizaje y la memoria.
El péptido Aβ está relacionado con la EA y es el principal componente de las placas amiloideas. El Aβ es el producto de la digestión proteolítica de la proteína precursora del amiloide (PPA). El gen que codifica a la PPA se encuentra en el brazo largo del cromosoma 21, y es una proteína integral de membrana, que está expresada en muchos tipos de tejidos, pero se concentra en la sinapsis entre neuronas.
Hoy en día se sabe que mutaciones en el gen de la PPA son responsables de 5 a 20% de los casos de la EA familiar precoz. Cabe destacar que la región de los residuos 21-23 del péptido Aβ se considera un “sitio caliente” para las mutaciones, debido al gran número de variantes genéticas reportadas en esta área de la molécula. Estas mutaciones poseen efectos neurotóxicos directos o exacerban los efectos dañinos de otras lesiones neurotóxicas.
Los procesos enzimáticos responsables del metabolismo de PPA para formar los Aβ se han ido esclareciendo con el paso de los años. Ahora se sabe que después de que la PPA es sintetizada, ésta es transportada a la membrana celular. Ahí es cortada secuencialmente por proteínas (las α-, β-, y γ- secretasas), con actividad de endoproteasa, que se encuentran ancladas en la membrana (Ver la Figura 2).
La actuación inicial de la α-secretasa sobre la PPA da lugar a la formación de péptidos solubles, fácilmente digeribles. Esta vía, llamada no-amiloidogénica, la cual impide la formación del péptido Aβ ya que el corte por la α secretasa ocurre dentro de la secuencia del péptido amiloide.
En la vía amiloidogénica, la proteína Aβ1-42 es una estructura de alfa hélice que después de la escisión de la PPA, rápidamente se transforma en estructuras desordenadas en disolución. La proteína Aβ1-42 es la forma monomérica (no se puede unir con otras proteínas) y es soluble en agua en el caso de un ser humano sano. Estas proteínas juegan un papel fundamental en el desarrollo y funcionamiento del cerebro; ya que son esenciales para el transporte de nutrientes a las células cerebrales para mantener un cerebro saludable y una regulación cerebral; y pueden ayudar a los neurotransmisores en el cerebro, que ayudan al cerebro a comunicarse y enviar señales al cuerpo
En el caso de los pacientes con EA, Aβ1-42 tiende a formar un dímero soluble (dos proteínas unidas), un trímero (tres proteínas se unen), oligómeros (más de cuatro proteínas unidas) y una fibrilla insoluble (varias proteínas organizadas en grupos). con patrones estructurales uniformes). Por lo tanto, el ensamblaje de especies Aβ1-42 se clasifica en dos categorías: agregación amorfa soluble, agregados de proteínas sin estructuras superiores específicas; agregación fibrilar insoluble, y agregados de proteínas con estructuras β cruzadas comunes.
Conjeturas sobre la enfermedad de Alzheimer
En 1901, el Dr. Alois Alzheimer, un psiquiatra alemán, observó comportamientos anormales en uno de sus pacientes de 51 años, a saber, Auguste Deter, que había sufrido graves problemas de memoria y no recordaba el nombre de su marido. Después de su muerte el 8 de abril de 1906, el médico identificó que su cerebro se había encogido significativamente ya que morían más neuronas por la formación de placas, ovillos, y la pérdida de conexiones entre neuronas. El médico caracterizó las condiciones patológicas de demencia antes mencionadas en el cerebro de su paciente; así, la enfermedad lleva su nombre, Enfermedad de Alzheimer. Los ovillos son estructuras anormales localizadas en varias partes del cerebro, principalmente dentro de las neuronas, y compuestas de agrupaciones densas de filamentos helicoidales (hoy en día se les conoce como ovillos neurofibrilares) apareados asociados a la proteína presente en los microtúbulos intracelulares llamada proteína tau. La proteína tau es un polipéptido que se encuentra preferentemente en los axones. La proteína tau experimenta una serie de cambios en la enfermedad de Alzheimer y se deposita en el cuerpo neuronal en vez de en los axones.
En 1992, Hardy y Higgins [1] desarrollaron la hipótesis de la “cascada amiloide”. Propusieron que los agregados de Aβ se transforman en fibrillas de Aβ, y que éstas se acumulan en el cerebro desencadenando finalmente la neurodegeneración. Cómo se mencionó anteriormente, el Aβ se produce a partir de la PPA transmembranal después de la escisión por las enzimas β- y γ- secretasas. La agregación de las proteínas Aβ ocurre fuera y alrededor de las neuronas en el cerebro llamadas placas amiloideas.
Por otro lado, en el cromosoma 17 se encuentra el gen que codifica la síntesis de la proteína tau, la cual es un polipéptido que se encuentra preferentemente en los axones. El papel de tau es proporcionar una estructura ‑‑como una especie de vía de tren‑‑ dentro de las neuronas del cerebro que permite a las células limpiar la acumulación de proteínas no deseadas y tóxicas. En la EA y otras enfermedades neurodegenerativas se produce una fosforilación irreversible de esta proteína que compromete su función normal y produce daño neuronal. Durante este proceso, la proteína tau experimenta una serie de cambios en la EA y se deposita en el cuerpo neuronal en vez de en los axones. siendo el integrante principal de los ovillos neurofibrilares (Ver la Figura 2).
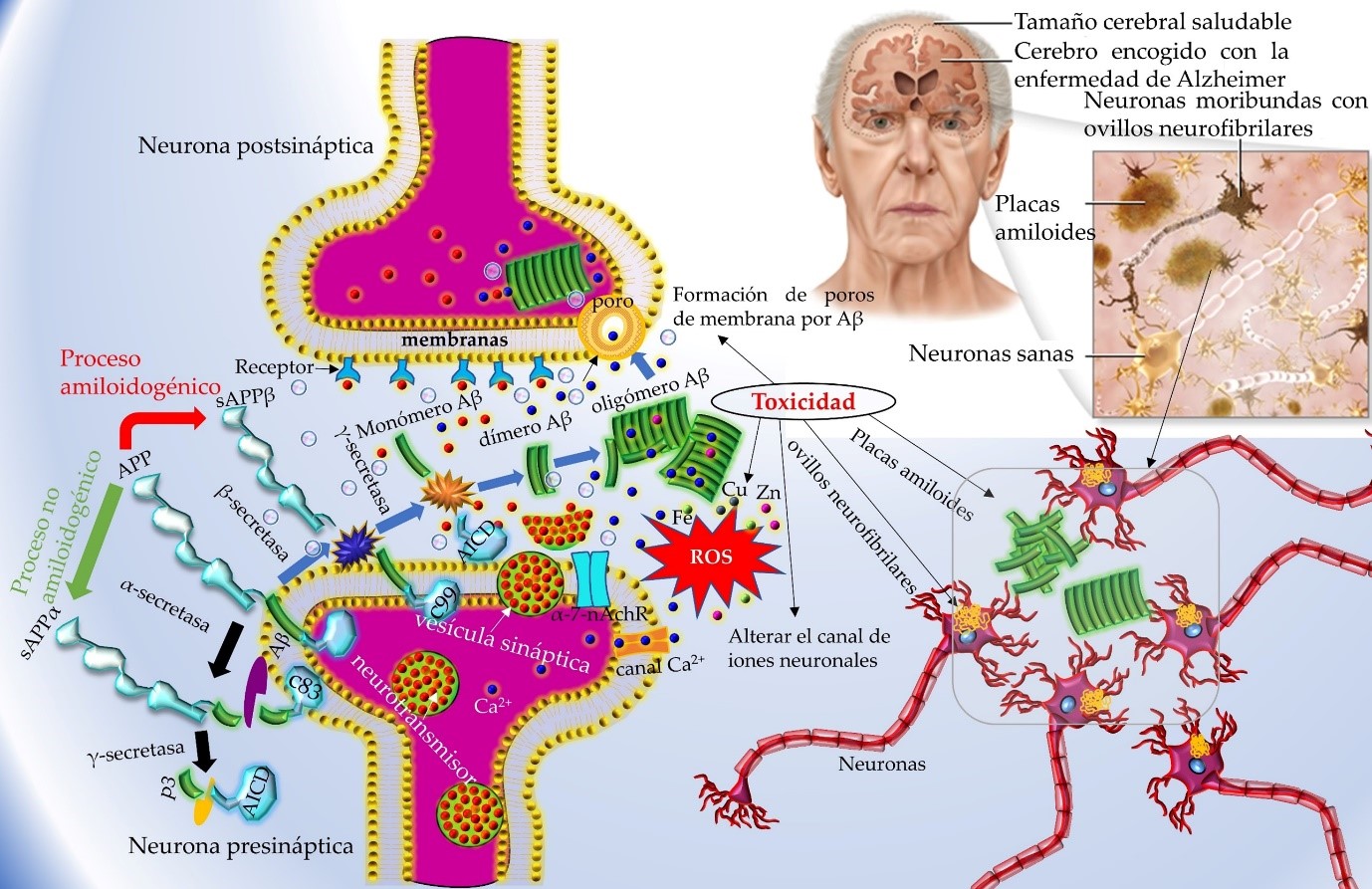
Figura 2: Hipótesis de placas amiloides y ovillos neurofibrilares (Adaptada de https://www.auhs.edu/wp-content/uploads/2020/07/Alzheimerpx.png).
Aunque cientos de moléculas pequeñas están presentes en el cerebro humano, los científicos han planteado la hipótesis de que las placas de amiloide y los ovillos neurofibrilares son los principales culpables del desarrollo de la enfermedad. Sin embargo, el mecanismo exacto de formación de la enfermedad de Alzheimer sigue siendo un misterio a pesar de que ya que pasaron más de cien años desde que se identificó la enfermedad. Además, no existe cura para la EA y la mayoría de los fármacos disponibles solamente reducen los síntomas de la enfermedad, por lo que el tratamiento es básicamente paliativo.
Dado que el aumento de la edad puede reducir la actividad de la enzima α-secretasa relacionada con la ralentización de la eliminación de la proteína Aβ en el cerebro, la enzima β-secretasa puede valerse de esto para producir proteínas Aβ enriquecidas. En un cerebro sano, las enzimas de degradación del amiloide (EDA), se invocan en el proceso de eliminación de proteína Aβ y regulan los niveles de proteína en el cerebro.
Además de la α-secretasa, otros estudios han identificado a la neprilisina como una proteasa involucrada en la degradación del péptido Aβ; asimismo, se ha reportado que la enzima degradadora de insulina (EDI), y las enzimas convertidoras de endotelina (ECEs), entre otras, también degradan este neuropéptido. Colectivamente, estas enzimas reciben el nombre de enzimas degradadoras de amiloide (EDA) y han generado un gran interés como potenciales candidatos para el tratamiento de la EA [2].
La EDI, también conocida como insilusina, es una metalopeptidasa dependiente de Zn que pertenece a la familia M16 de las metaloproteínas. La EDI se distribuye ubicuamente en el organismo, pero es abundante principalmente en músculos, hígado y cerebro. Esta enzima fue inicialmente identificada por su capacidad de degradar la insulina, pero actualmente se reconoce un grupo más amplio de sustratos [3]. Un estudio reciente encontró que EDI reconoce el sitio Met (35)-Val (36) en la secuencia del péptido Aβ, produciendo fragmentos proagregantes amorfos menos tóxicos que el péptido Aβ [4].
Con el envejecimiento el nivel y la actividad de la EDA y α-secretasa disminuyen, y la edad avanzada favorece la formación de placas y, por lo tanto, las personas mayores tienen un mayor riesgo de desarrollar EA.
Importancia de los iones metálicos en la EA
Se ha observado que los cerebros post mortem de los pacientes con EA tienen 1 mM de zinc (Zn2+), 0,4 mM de cobre (Cu2+) y 1 mM de hierro (Fe3+), que son 3.1, 5.7 y 2.8 veces más altas que un cerebro sano. Estas concentraciones deben ser muy variables, a veces alguno de los iones estará más alto que los otros, etc. Es necesario revisar la información o no dar datos puntuales, sino intervalos. Se ha comprobado que estos iones metálicos pueden contribuir significativamente a la formación de placas. Por ejemplo, la mayor concentración de iones metálicos Cu2+ y Zn2+ inhibe la fibrilación de Aβ1-42 y promueve la agregación amorfa. Después, los agregados amorfos se convierten en una fibrilla mediante la unión de iones metálicos. Esta evidencia refleja que los agregados amorfos no son el punto final en las vías de agregación.
En el caso del cerebro sano, la concentración promedio de Cu2+ y Fe3+ están generando especies de oxígeno redox (ROS) que juegan un papel importante en la función cerebral, como el metabolismo oxidativo, la plasticidad sináptica, la mielinización y la generación de neurotransmisores. Mientras que, en el caso de los cerebros de los pacientes con EA, esos iones metálicos alcanzan una concentración más alta, produciendo ROS reactivas enriquecidas por reacciones similares a las de Fenton (una oxidación avanzada en el cual se producen radicales altamente reactivos del hidroxilo), lo que resulta en un deterioro neuronal desencadenado por la agregación del oligómero Aβ tóxico, lo que provoca defectos cognitivos [5].
Las proteínas Aβ pueden reducir Cu2+ a Cu+, y Fe3+ a Fe2+, lo que facilita la generación de especies reactivas de oxígeno H2O2 y radicales OH•. Las ROS pueden eliminar los iones de hidrógeno de la membrana celular y la peroxidación de lípidos en la membrana neuronal, lo que provoca la muerte neuronal y la disfunción orgánica. En particular, dos residuos se oxidan en Aβ, Met35 se oxida a un sulfóxido y Tyr10 se oxida para formar radicales de ditirosina inducidos por oligómeros de Aβ tóxicos. Las ROS enriquecidas y el estrés oxidativo (desequilibrio entre oxidantes y antioxidantes en el sistema biológico) pueden desencadenar la producción de Aβ al afectar o reducir la actividad de las enzimas α-secretasa en PPA [6].
En 1994, Khachaturian [7] imaginó la "hipótesis del calcio en el envejecimiento cerebral" que describe a los iones Ca2+ como un elemento indispensable para la función cerebral que contribuye a la liberación de neurotransmisión, la plasticidad sináptica y la expresión génica.
Recientemente se ha descubierto que las neuronas del córtex prefrontal desarrollan una fuga en el almacenamiento de calcio con la edad. Esta interrupción del almacenamiento de calcio conduce, a su vez, a la acumulación de proteínas tau fosforiladas o modificadas, causantes de los ovillos neurofibrilares distintivos del Alzheimer [8]. Este efecto interviene en las disfunciones cognitivas al generar neuroinflamación, falla sináptica, neurotoxicidad y plasticidad sináptica. Por lo tanto, la relación entre Aβ y los iones Ca2+ mediaron los déficits cognitivos en pacientes con EA [9].
En otras palabras, en el caso del cerebro sano, el flujo de iones de calcio en la membrana puede aumentar la liberación de vesículas sinápticas, y de los neurotransmisores almacenados en ellas, en las neuronas del hipocampo. Sin embargo, Aβ puede reducir significativamente el número de vesículas que fallan en la neurotransmisión. Además, las proteínas Aβ pueden interactuar directamente con la membrana neuronal formando una estructura de poros en la que el flujo de calcio desencadena la muerte celular. Esto último está bien, pero requiere una referencia.
Enfermedad de Alzheimer familiar
La forma ártica de EA es una variante de enfermedad rara, la cual está ligada a una mutación (E669G o E22G) dentro de la región amiloidea del gen de la PPA. Esta mutación se identificó en una familia de cuatro generaciones del norte de Suecia. Los individuos afectados presentaban características clínicas de la enfermedad de Alzheimer de aparición temprana con una edad media de aparición de 57 ± 2.9 años y deterioro cognitivo insidioso.
La forma ártica de EA aumenta las especies de oligómeros Aβ modificados de tipo salvaje y mutados que se propagan de manera extracelular e intracelular en las neuronas [10]. Los oligómeros extracelulares no dañan a las neuronas ni inducen la formación de ovillos neurofibrilares; en contraste, los oligómeros de Aβ intracelulares son más neurotóxicos que los depósitos de Aβ extracelulares. Además, los depósitos intracelulares de Aβ favorecen el enriquecimiento de las marañas neurofibrilares, lo que dificulta el transporte axoplásmico y contribuye a la pérdida sináptica.
Las neuronas humanas acumulan preferentemente Aβ1-42 sobre Aβ1-40, ya que estudios in vitro que muestran que las proteínas extendidas en el C terminal tienen una tendencia mucho mayor a formar una fibrilla de amiloide. Por otro lado, la mutación ártica en las proteínas Aβ1-42 promueve la formación de proteínas prefibrilares más rápido que la Aβ1-42 salvaje, así como en la mutación ártica de Aβ1-40, que es el mecanismo molecular detrás de la forma ártica del desarrollo de la enfermedad de EA. La mutación ártica es bastante peligrosa entre otras mutaciones, ya que ha inducido una mayor toxicidad [11].
Medicinas para la enfermedad de Alzheimer
Existen dos tipos de moléculas de fármacos inhibidores de la colinesterasa y memantina. Los inhibidores de la colinesterasa pueden aumentar la cantidad de colinesterasa, que es responsable de las funciones de la memoria. La memantina regula la actividad del glutamato, que es un químico mensajero involucrado en la función cerebral, incluyendo el aprendizaje y la memoria. Normalmente, el mediador importante es la acetilcolina y la colinesterasa la destruye en la sinapsis por lo que estos fármacos la hacen estar activa más tiempo.
Existen tres inhibidores de la colinesterasa: el donepezil (Aricept), la galantamina (Razadyne) y la rivastigmina (Exelon). El donepezil se usa en todas las etapas de los pacientes con EA, la galantamina sirve para tratar la enfermedad de leve a moderada y la rivastigmina ayuda en el estado grave de los pacientes con EA.
No sabemos la etiología de la EA ya que a la fecha no existe un tratamiento específico, es necesario seguir investigando.
Cabe destacar que, el 7 de junio de 2021, la FDA de los EUA aprobó una nueva molécula de fármaco, el aducanumab, para pacientes con EA leve debido a que facilita la eliminación de las placas ricas en amiloides y ha minimizado los efectos secundarios. Hay que tener en cuenta que todas las moléculas de fármacos discutidas anteriormente alivian a los pacientes con EA, pero no curan la enfermedad porque hay dos obstáculos principales: (1) Aunque existen cientos de moléculas pequeñas en el cerebro humano, los ensayos clínicos se dirigen a dos proteínas, Aβ y tau, y (2) Los estudios experimentales no pueden imitar exactamente la membrana cerebral y preparar oligómeros de Aβ, y las estructuras de las fibrillas no se parecen a las que se presentan en los cerebros de los pacientes con EA [12]. Al final, a pesar de que hay varios medicamentos contra la EA disponibles en el mercado, ninguno de ellos cura la enfermedad por completo.
Por todo lo expuesto, la investigación en un tema tan relevante para la humanidad es de gran importancia y hay muchos grupos trabajando en ello. Sin duda habrá avances.
Referencias
- Hardy, J. A., & Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science, 256(5054), 184-185. https://doi.org/10.1126/science.1566067
- Nalivaeva NN, Belyaev ND, Kerridge C, Turner AJ. Amyloid-clearing proteins and their epigenetic regulation as a therapeutic target in Alzheimer's disease. Front Aging Neurosci. 2014; 6:235. https://doi.org/10.3389/fnagi.2014.00235
- Hersh LB. The insulysin (insulin degrading enzyme) enigma. Cell Mol Life Sci. 2006;63(21):2432-4. https://doi.org/10.1007/s00018-006-6238-9
- Hubin, E., Cioffi, F., Rozenski, J., Van Nuland, N. A., & Broersen, K. (2016). Characterization of insulin-degrading enzyme-mediated cleavage of Aβ in distinct aggregation states. Biochimica et Biophysica Acta (BBA)-General Subjects, 1860(6), 1281-1290. https://doi.org/10.1016/j.bbagen.2016.03.010
- Boopathi, S.; Poma, A. B.; Garduño-Juárez, R. An Overview of Several Inhibitors for Alzheimer’s Disease: Characterization and Failure. Int. J. Mol. Sci. 2021, 22 (19), 10798. https://doi.org/10.3390/ijms221910798
- Boopathi, S.; Kolandaivel, P. Fe2+ Binding on Amyloid β-Proteins Promotes Aggregation. Proteins Struct. Funct. Bioinforma. 2016, 84 (9), 1257–1274. https://doi.org/10.1002/prot.25075
- Khachaturian, Z. S. (1994). Calcium Hypothesis of Alzheimer's Disease and Brain Aging. Annals of the New York Academy of Sciences, 747(1), 1-11. https://doi.org/10.1111/j.1749-6632.1994.tb44398.x
- Datta, D., Leslie, S. N., Wang, M., Morozov, Y. M., Yang, S., Mentone, S., ... & Arnsten, A. F. (2021). Age‐related calcium dysregulation linked with tau pathology and impaired cognition in non‐human primates. Alzheimer's & Dementia, 17(6), 920-932. https://doi.org/10.1002/alz.12325
- Thibault, O., Gant, J. C., & Landfield, P. W. (2007). Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging cell, 6(3), 307-317. https://doi.org/10.1111/j.1474-9726.2007.00295.x
- Kalimo, H., Lalowski, M., Bogdanovic, N., Philipson, O., Bird, T. D., Nochlin, D., ... & Ingelsson, M. (2013). The Arctic AβPP mutation leads to Alzheimer’s disease pathology with highly variable topographic deposition of differentially truncated Aβ. Acta neuropathologica communications, 1(1), 1-19. https://doi.org/10.1186/2051-5960-1-60
- Lu, M., Williamson, N., Mishra, A., Michel, C. H., Kaminski, C. F., Tunnacliffe, A., & Schierle, G. S. K. (2019). Structural progression of amyloid-β Arctic mutant aggregation in cells revealed by multiparametric imaging. Journal of Biological Chemistry, 294(5), 1478-1487. https://doi.org/10.1074/jbc.RA118.004511
- Knoblich, J. A. (2017). Lab-built brains. Scientific American, 316(1), 26-31. https://doi.org/10.1038/scientificamerican0117-26
Esta columna se prepara y edita semana con semana, en conjunto con investigadores morelenses convencidos del valor del conocimiento científico para el desarrollo social y económico de Morelos. Desde la Academia de Ciencias de Morelos externamos nuestra preocupación por el vacío que genera la extinción de la Secretaría de Innovación, Ciencia y Tecnología dentro del ecosistema de innovación estatal que se debilita sin la participación del Gobierno del Estado.
Más publicaciones