Origami molecular y el premio Nobel de química 2024

Escrito por:
Dra. Nina Pastor Colón,
Dr. Enrique Rudiño Piñera
Visto: 3618 veces
Origami molecular y el premio Nobel de química 2024
Nina Pastor Colón y Enrique Rudiño Piñera
La Dra. Nina Pastor es profesora-investigadora del Centro de Investigación en Dinámica Celular de la Universidad Autónoma del Estado de Morelos. Su área de especialidad es la biofísica molecular computacional, con énfasis en el estudio del reconocimiento entre moléculas. Es integrante de la Academia de Ciencias de Morelos.
El Dr. Enrique Rudiño es investigador titular del Instituto de Biotecnología de la Universidad Nacional Autónoma de México (UNAM) y responsable del grupo de Bioquímica Estructural del Instituto de Biotecnología de la UNAM en Cuernavaca, Morelos. Es integrante de la Academia de Ciencias de Morelos.
Esta publicación fue revisada por el comité editorial de la Academia de Ciencias de Morelos.
Este año el premio Nobel de química se otorgó a Demis Hassabis y John Michael Jumper por su trabajo sobre la predicción de la estructura de las proteínas y a David Baker por enfoques novedosos para diseño de proteínas. Para entender por qué estos avances merecen un premio Nobel, los invitamos a leer la siguiente historia.
El plegamiento de proteínas como un ejercicio de origami molecular
El arte japonés de doblar papel para generar figuras únicas se parece mucho al proceso mediante el cual las proteínas adquieren una forma o plegamiento (Figura 1). Imaginemos a una proteína como una tira larga de papel, con segmentos de veinte colores distintos (cada color sería un aminoácido, los bloques que al unirse dan lugar a una proteína particular); esta tira no parecerá una grulla o mariposa sin antes doblarse sobre sí misma, a veces generando incluso nudos, para dar lugar a una escultura en tres dimensiones. Producto del proceso de plegamiento o doblado, distintas secciones de la tira de papel, que cuando está estirada están muy lejos entre sí, quedan cerca unas de otras, generando cuevas, riscos y plataformas útiles para unirse con otras tiras de papel (otras proteínas) o incluso unirse con objetos distintos, como si a esa tira de papel le pegáramos una pieza de chaquira en un lugar específico (otras moléculas).
 Figura 1: Muestras de origami https://images.app.goo.gl/3s4AMgX8ikiUy4cP9
Figura 1: Muestras de origami https://images.app.goo.gl/3s4AMgX8ikiUy4cP9
En las proteínas no hay un pegamento como el que usamos para estabilizar un doblez particular; en lugar de eso existen las interacciones moleculares que se forman entre los distintos tipos de aminoácidos; hay unos con carga positiva, otros con carga negativa, otros que interactúan bien con el agua, y otros que son aceitosos. La conformación o doblez más estable es aquella en la que los vecinos de cada aminoácido contribuyen a generar un ambiente favorable para interactuar con otros. Los que interactúan bien con el agua estarán normalmente en la superficie de la proteína, generalmente rodeada de mucha agua. Los aminoácidos aceitosos casi siempre buscarán estar escondidos del agua, en el interior de la estructura de la proteína en cuestión. La forma, o estructura, de cada proteína conocida contiene pistas acerca de su función, siguiendo la máxima biológica de la relación entre la estructura y la función. Es como si observáramos un serrucho y no supiéramos para que se usa, pero al analizar su forma podríamos intuir que se trata de una herramienta para cortar. Esto aplica también a nivel molecular.
En algunas cavidades formadas al plegar la tira de papel se juntarían segmentos, que en las proteínas llamamos grupos químicos; éstos serían capaces de ayudar a otras moléculas a transformarse de una manera mucho más eficiente que en su ausencia. Esto define a lo que llamamos catalizadores, tan específicos que distinguen la quiralidad de las moléculas y la posición específica del mismo grupo funcional en distintos sitios de las moléculas; esto permite salvar vidas, reducir costos, biorremediar aguas, y producir medicamentos, entre muchas otras cosas. Las cuevas o cavidades se llaman sitios activos en las proteínas, por tener actividad química. Las cuevas, riscos y plataformas sin actividad química se llaman sitios de unión, y son importantes para la sociología molecular, ya que las proteínas funcionan interactuando con ellas mismas o con otras moléculas presentes en su entorno, como lo hacemos los humanos.
Christian Anfinsen (premio Nobel en 1971) encontró que las instrucciones para plegar una proteína se encuentran en la secuencia de aminoácidos de cada proteína. Mostró que la estructura en la cual se pliegan en condiciones normales o nativas (aquellas congruentes con su función biológica), es siempre la misma. Eso implica que esa estructura corresponde al mínimo de energía libre del sistema, concepto poderoso porque si pudiéramos determinar la energía libre de cada una de las maneras posibles de plegar una proteína (o a la tira de papel), podríamos elegir cuál es el plegamiento funcional.
Desde principios del siglo XXI sabemos que existe una clase de proteínas que no adoptan una estructura nativa única, sino muchas. Éstas son las proteínas intrínsecamente desordenadas, o IDPs por sus siglas en inglés. En este caso hay muchas estructuras o plegamientos de la tira de papel con energías similares, por lo que no hay una manera de elegir “la mejor”: hay muchas igual de buenas. Los físicos estadísticos llaman a este tipo de sistemas “frustrados”, porque no encuentran una manera de estar completamente a gusto, y se la pasan cambiando de postura en una danza infinita.
En 1969 Cyrus Levinthal hizo un estimado rápido (lo que los físicos llaman cálculos de servilleta o de sobre), de cuánto tiempo necesitaría una proteína pequeña, de sólo 100 aminoácidos, para encontrar su conformación nativa. Para entender este cálculo, imaginen a nuestra tira de papel como una cadena de pedacitos de papel unidos por anillos que les permiten girar a unos respecto a otros. En la figura 2 los papeles serían los eslabones que vemos de frente, y los anillos, los que se ven de perfil.

Figura 2: Fotografía de una cadena
En una cadena de aminoácidos, los anillos son los carbonos alfa, los cuales conectan a los enlaces peptídicos entre un aminoácido y el siguiente. Estos enlaces peptídicos son planos y rígidos (serían los pedacitos de papel). Para hacer las cuentas más fáciles, Levinthal consideró que cada anillo podia estar en nueve posiciones distintas, tres a cada lado del carbono alfa. Si la cadena es de 100 aminoácidos, tenemos un total de 9100 arreglos posibles; este número es astronómicamente grande. Ahora consideremos que la rotación de estos enlaces toma unos 10-12 segundos (una millonésima de una millonésima de segundo, o un picosegundo). Aún girando así de rápido, resulta que a una proteína de 100 aminoácidos le tomaría más que la edad del universo para encontrar su conformación nativa. Las proteínas encuentran su estado nativo en fracciones de segundos o a lo más en unos minutos, por lo que una búsqueda sobre todos los arreglos posibles en el espacio no es lo que hacen. La solución es similar a seguir las instrucciones de un manual de origami: hay un orden preciso para plegarse (o una colección pequeña de caminos para llegar al sitio correcto). ¡Este orden también está incluído en la secuencia de aminoácidos de las proteínas, y se conoce como la o las rutas de plegamiento!
El problema del plegamiento de las proteínas y el problema de la predicción de estructuras de proteínas
Hay proteínas que se pliegan reproduciblemente a una estructura particular, la nativa y funcional, y que lo hacen en tiempos biológicamente razonables (segundos o minutos). También hay otras proteínas que tienen muchas estructuras nativas, todas similarmente probables (las IDPs). En ambos casos, la secuencia de aminoácidos dicta su comportamiento. Conocemos las estructuras tridimensionales de muchas proteínas plegadas, gracias al esfuerzo comunitario de los biólogos estructurales del mundo, quienes han depositado estas estructuras en el Protein Data Bank (PDB: https://www.rcsb.org) [1], un repositorio abierto muy recomendable con su portal educativo (https://pdb101.rcsb.org). En esta base de datos no existen estructuras de IDPs, ya que no se están quietas en una sola estructura. Para las IDPs una sola foto no tiene sentido; hay que describirlas usando herramientas de la física de polímeros, mirando la colección de todas las estructuras que pueden adoptar en un periodo de tiempo.
Es muchísimo más fácil determinar las secuencias de aminoácidos de las proteínas que determinar su estructura tridimensional, y eso se nota al comparar el tamaño de las bases de datos que albergan secuencias de aminoácidos con el PDB. Las primeras tienen muchos millones de secuencias, mientras que el PDB tiene apenas unas 200,000 estructuras. Esta disparidad va a seguir creciendo, por lo que a los bioquímicos y biólogos moleculares nos interesa sobremanera saber con la mayor precisión posible, nada más con ver la secuencia de aminoácidos, cómo se va a plegar en tres dimensiones, porque de la forma depende la función.
Este problema se puede resolver de dos maneras. Una es usando conocimientos de la física de las interacciones entre aminoácidos y el agua, y buscando el mínimo de energía libre; así puede llegarse al estado nativo y entender el proceso de plegamiento. Con este enfoque, útil sólo para proteínas pequeñas, se han logrado plegamientos in silico, pero esto requiere a las súpercomputadoras dedicadas como Anton o el cómputo distribuido de Folding@Home [2,3]. Para proteínas de tamaño mayor, la opción es abandonar la ambición de entender el mecanismo y “simplemente” predecir la forma final. La mitad del premio Nobel de química 2024, dado a Demis Hassabis y John Michael Jumper por el desarrollo de AlphaFold2, está dedicado a la segunda manera de enfrentar el problema del plegamiento de proteínas. No resuelve el problema del mecanismo de plegamiento, pero sí propone estructuras de proteínas a partir de la secuencia de aminoácidos, en tiempos cortos y con un indicador de la calidad de la predicción local, para cada aminoácido de la cadena.
¿Qué hace a AlphaFold2 tan bueno para predecir estructuras de proteínas?
Aquí se vuelve a aplicar el aforismo de se puede ver más lejos al estar parado sobre hombros de gigantes. Una buena parte del éxito del programa AlphaFold2 es que utiliza como material de entrenamiento las estructuras de proteínas conocidas, depositadas en el PDB. Con eso, el programa aprende cómo se ve una proteína decentemente plegada (qué va en el centro, escondido del agua, y qué va afuera, expuesto al agua, o a cuáles aminoácidos les gusta estar juntos, por ejemplo). La otra mitad de la información está en la secuencia de aminoácidos de la proteína para la cual se quiere conocer la estructura. Para entender esto necesitamos información de la evolución.
Todos los organismos del planeta somos descendientes de un conjunto primordial de seres vivos conocidos como LUCA (último ancestro común universal). Como consecuencia, las proteínas a cargo de muchos procesos biológicos son tan viejas como la vida en nuestro planeta (del orden de 3800 millones de años), y que conforme se han heredado han ido sufriendo pequeñas modificaciones en su secuencia de aminoácidos, pero siguen plegándose de la misma manera y realizando, casi siempre, la misma función. Por lo tanto, si yo pregunto en una base de datos de secuencias de aminoácidos si mi proteína favorita tiene familiares (si hay secuencias de aminoácidos a las que se parezca), puedo construir algo llamado un alineamiento de secuencias. En cada renglón de este alineamiento pongo la secuencia de aminoácidos de las proteínas que se parecen entre sí, con la mía hasta arriba, acomodadas de manera que las columnas coincidan lo más posible. Hacer un alineamiento implica que, si miro una columna en el alineamiento, la posición de ese aminoácido en el espacio es igual en todas las proteínas alineadas. Esto es una idea poderosa porque en general, las posiciones clave para mantener la arquitectura de la proteína se conservan a lo largo de la evolución (no cambia el aminoácido, aunque estemos comparando arqueas con humanos). Cuando hay cambios puede suceder que estén correlacionados entre varias posiciones: si se imaginan dos aminoácidos frente a frente, uno grande y uno chico, o uno positivo y uno negativo, lo que suele verse es que si el grande cambia a uno chico, el chico que tenía enfrente cambia a uno grande; si era una pareja cargada, si el positivo se vuelve negativo, el negativo se vuelve positivo; es como una pareja danzando el que el movimiento de un bailarín produce un efecto en el movimiento del otro. Por lo tanto, examinando en el alineamiento cuáles columnas cambian de manera correlacionada se puede adivinar cuáles aminoácidos podrían estar próximos en la estructura tridimensional final.
Ahora ya tenemos lo esencial para el trabajo de AlphaFold2: una idea de cómo se ve una proteína plegada y una adivinanza inicial de cuáles aminoácidos de mi proteína favorita deberían de estar cerca. El juego consiste en encontrar estructuras que satisfagan simultáneamente la mayor cantidad posible de contactos esperados a partir del alineamiento, sujetos a la restricción de que los aminoácidos están conectados entre sí en una cuerda (la tira de papel), y a las reglas aprendidas sobre cómo se ve tridimensionalmente una proteína. Esto se hace de manera iterativa, y al final del proceso AlphaFold2 nos da cinco respuestas razonables de estructuras para la secuencia de nuestra proteína favorita. Dependiendo de su grado de confianza, colorea a los aminoácidos desde un color azul oscuro (alta confianza), azul claro (mediana confianza), amarillo (poca confianza), naranja (muy poca confianza) y rojo (no me creo nada, pero tú lo pediste). Si algún lector quiere intentarlo, existe un enlace para realizar gratuitamente las predicciones que uno quiera: https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb
Hay dos casos en los cuales AlphaFold2 falla. Uno de ellos son las IDPs. Como ejemplo de lo anterior, la figura 3 muestra la predicción hecha para una proteína llamada Escargot. Esta proteína tiene una sección ordenada que usa para unirse al ADN, y que requiere zinc para plegarse. Corresponde a los listones azul claro en la estructura. Buena parte del resto de la estructura está desordenada, y eso se nota en los listones de color amarillo o naranja, los cuales recuerdan más a un plato de espaguetis. Si se fijan, los conectores entre las regiones azules también están en amarillo y naranja, lo cual implica que no se puede decir nada serio sobre su posición relativa. Hay muchas estructuras predichas por AlphaFold2 que se ven así … y en nuestra opinión, no podemos hablar de una predicción de estructura con tantas regiones sobre las que no se puede decir nada.
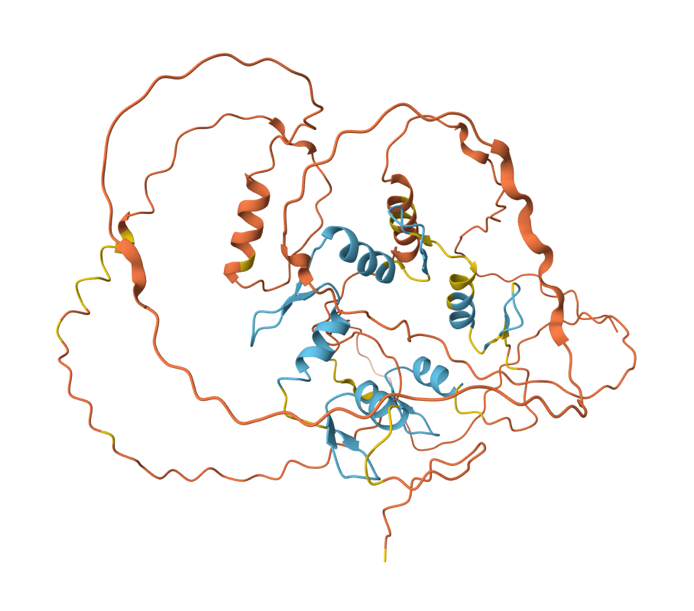
Figura 3: Predicción de AlphaFold2 para la estructura de la proteína Escargot de Drosophila melanogaster (registro AF-P25932-F1). Las zonas predichas con confianza se muestran en azul.
El otro caso en el que AlphaFold2 falla se da cuando nuestra proteína favorita no tiene muchos familiares. En ese caso el alineamiento tiene muy pocos miembros, y entonces es difícil conseguir un número grande de correlaciones que se traduzcan en distancias estimadas para distintas parejas de aminoácidos. Si además de eso nuestra proteína favorita requiere la formación de puentes disulfuro para ser estable (enlaces realizados entre los átomos de azufre del aminoácido cisteína), eso complica más la predicción, porque estos puentes suelen conservarse y no tienen cambios correlativos.
Aún en condiciones ideales de trabajo, dada la manera de proponer estructuras por parte de AlphaFold2 (comienza con un “gas” de aminoácidos, no una cadena), los detalles de estereoquímica de la proteína suelen ser incorrectos. Como ya se conoce el problema, al resultado de AlphaFold2 se le da una manita de gato que consiste en una minimización de energía, con lo cual se recupera la estereoquímica. Otra cosa, que no se arregla con la minimización, es la predicción de la conformación de las cadenas laterales de aminoácidos, sobre todo los que están en la superficie de la proteína, los cuales son particularmente difíciles. Uno pudiera pensar que esto es menor, total, están expuestas al solvente y se van a mover, pero es un problema serio si uno quiere hacer experimentos de acoplamiento molecular directamente con la propuesta de AlphaFold2, con el fin de proponer un nuevo fármaco por ejemplo. Pudiera ser que las plataformas, riscos y cuevas no tengan la conformación adecuada para unirse al ligando de interés. Eso se arregla realizando simulaciones de dinámica molecular, para tener una colección de poses para la superficie, y sobre esa colección se hace el acoplamiento molecular.
Bajo las condiciones óptimas de operación, AlphaFold2 es una herramienta excelente para la predicción de estructura de proteínas, y es un punto de partida muy bueno para hacer química medicinal. Sin embargo, es crucial revisar con cuidado el resultado. Como casi todo en la vida, la verdad esta escondida en los detalles. Es recomendable incluir en la predicción solamente los aminoácidos presentes en la proteína madura, cosa que no sucede en las predicciones automáticas depositadas en bases de datos. Por último, si lo que a uno le interesa es una sección que el programa pinta de amarillo, naranja o rojo … es mejor no usar el modelo.
Viendo el problema en la dirección opuesta: la otra mitad del premio Nobel
Hasta ahora hemos platicado sobre la predicción de una estructura dada una secuencia de aminoácidos. La otra cara de la moneda es preguntar, dada una estructura particular, cuál sería la secuencia de aminoácidos compatible con ella. El experto mundial en esto es David Baker, galardonado con la otra mitad del premio Nobel de química 2024, por sus contribuciones al diseño de proteínas.
Además de competir efectivamente en el tema de la predicción de estructura con su servidor Robetta (https://robetta.bakerlab.org ), David Baker y su grupo imaginan proteínas que no existen o no hemos encontrado aún en la naturaleza. También tienen un juego llamado Foldit (https://fold.it), con el cual uno puede aprender a plegar proteínas inmerso en un juego de video. Foldit es una iniciativa de ciencia ciudadana, y en su página se ponen problemas abiertos de diseño para que la gente juegue con el programa, y de paso contribuya a resolver los problemas que permiten a la ciencia de proteínas seguir avanzando.
La primer proteína diseñada con un plegamiento completamente novedoso se llama Top7. Para construirla, primero imaginaron el trazo de la proteína en el espacio (la tira de papel), y después colorearon la tira de papel, lo cual se traduce en colocar las cadenas laterales de los aminoácidos de forma que se empacaran bien en el corazón de la proteína y la superficie fuera soluble en agua. Teniendo la secuencia de aminoácidos, sintetizaron el gene que la codifica, se lo dieron a una bacteria, y la bacteria produjo la proteína. Al purificar y cristalizar a Top7, obtuvieron la estructura experimentalmente, que resultó ser básicamente la que se predijo desde el inicio del experimento. Esta proteína no “sirve” para nada, pero es una prueba de concepto impresionante: estamos en la antesala del diseño de proteínas para funciones específicas e importantes.
Lo siguiente fue diseñar proteínas con un propósito en particular, como el diseño de una enzima que hace la reacción del Diels-Alder (una reacción en la que dos moléculas con dobles enlaces se unen en ciclos con estereoquímica precisa): conociendo la química de la reacción, organizaron aminoácidos alrededor del punto clave de la reacción (el estado de transición), y luego diseñaron el camino global de la proteína, de manera que los aminoácidos del sitio activo quedaran en la posición correcta. Este primer diseño produjo un catalizador modesto; para mejorarlo lo incluyeron como un problema en Foldit. Gracias a las contribuciones de muchos usuarios, lograron mejorar la actividad de esta proteína [4].
RoseTTAFold [5] tiene la capacidad de “alucinar” estructuras. Sí, el programa genera estructuras novedosas, para muchas de las cuales no hay una función aparente todavía. Además de esto, en el laboratorio de David Baker se han dedicado a diseñar proteínas con funciones específicas, como inhibidores de la infección por el SARS-CoV-2.
El premio Nobel de Química 2024 muestra, entre otras cosas, que la interrelación de la bioquímica y biofísica con la física y la ciencia computacional es un ecosistema vigoroso, en pleno crecimiento. Aprender sobre inteligencia artificial y aprendizaje de máquinas ya no es un lujo o algo para un nicho muy particular de investigadores, sino algo que tiene influencia clara en los aspectos más fundamentales de la biología: la estructura y función de las proteínas de todos los seres vivos del planeta.
Referencias:
[1] Berman HM et al. “The Protein Data Bank” Nucleic Acids Research 28:235-242 (2000)
[2] Lindorff-Larsen K et al. “How fast-folding proteins fold” Science 334: 517-20 (2011)
[3] Voelz VA et al. “Folding@home: Achievements from over 20 years of citizen science herald the exascale era” Biophysical Journal 122: 2852-2863 (2023)
[4] Eiben CB et al. “Increased Diels-Alderase activity through backbone remodeling guided by Foldit players” Nature Biotechnology 30: 190-192 (2012)
[5] Krishna R et al. “Generalized biomolecular modeling and design with RoseTTAFold All-Atom” Science 384: eadl2528 (2024)
Para saber más:
- ¿Las proteínas tienen una forma tridimensional? y eso ¿cómo afecta mi vida? Dr. Enrique Rudiño Piñera, 21 noviembre 2023 https://acmor.org/publicaciones/las-prote-nas-tienen-una-forma-tridimensional-y-eso-c-mo-afecta-mi-vida
- Doblando collares con pura intuición Dr. Gabriel del Río Guerra, 24 julio 2023 https://acmor.org/publicaciones/doblando-collares-con-pura-intuici-n
- Nota de prensa para todo público sobre el premio Nobel de química 2024: https://www.nobelprize.org/prizes/chemistry/2024/popular-information/
Esta columna se prepara y edita semana con semana, en conjunto con investigadores morelenses convencidos del valor del conocimiento científico para el desarrollo social y económico de Morelos.
Más publicaciones