¿Cómo surgen los virus que infectan al humano? Origen y evolución del SARS-CoV-2

Escrito por:
Dr. Alejandro Miguel Cisneros Martínez
Visto: 3832 veces
¿Cómo surgen los virus que infectan al humano? Origen y evolución del SARS-CoV-2
Alejandro Miguel Cisneros Martínez
Alejandro Cisneros es Biólogo y Maestro en Ciencias Biológicas por parte de la Facultad de Ciencias de la UNAM. Actualmente es estudiante del Doctorado en Ciencias Biomédicas en el grupo de Origen de la Vida dirigido por el Dr. Antonio Lazcano. Su trabajo se ha enfocado en usar a los virus de RNA como modelo para comprender los mecanismos de crecimiento y evolución de este tipo de genomas en etapas tempranas de la evolución de la vida.
Esta publicación fue revisada por el comité editorial de la ACMor.
Me gustaría empezar señalando que los virus son un fenómeno de la naturaleza íntimamente ligado a los seres vivos. Todos los seres vivos, desde el más pequeño hasta el más grande, son infectados por algún tipo de virus. Estas relaciones existen desde hace miles de millones de años, antes de que el ser humano existiera y en algunos casos, los virus podrían ser casi tan antiguos como la vida misma. Hay tantos virus en el mundo que se estima que, si todas las partículas virales se acomodaran en una cadena, esta se extendería por más de 200 millones de años luz en el espacio. Por eso, no debería sorprendernos que la humanidad sea frecuentemente azotada por infecciones virales. Los virus que empezaron a infectar al humano de manera más reciente son virus de RNA. Estos son virus cuya información hereditaria es almacenada en un tipo de molécula llamada ácido ribonucleico (o RNA por sus siglas en ingles), que es similar al DNA, molécula donde se almacena la información hereditaria en todos los seres vivos. Ejemplos de virus de RNA son el virus de la inmunodeficiencia humana (VIH), Ébola, Zika, Dengue, Chikunguya, los virus de la influenza y los coronavirus. Como se describe en otro artículo de esta columna (http://www.acmor.org/articulo/un-nuevo-coronavirus-que-es-eso-nos-tenemos-que-preocupar) la emergencia o surgimiento de estos virus como patógenos de humano se ha dado a través de un proceso natural conocido como zoonosis. Los virus de RNA están dotados con una extraordinaria capacidad de saltar de una especia a otra con gran facilidad debido a que tienen una alta tasa de mutación. Las mutaciones también son un proceso natural que le ocurren a todos los seres vivos y son consideradas como el combustible de la evolución. Por lo tanto, una alta tasa de mutación implica una rápida evolución (la evolución implica cambios en las especies que les permiten seguir existiendo). Por lo general, las infecciones virales suelen ser poco severas para el hospedero original pero muy severas para el nuevo hospedero. Sin embargo, gracias a la evolución, es posible que, con el paso del tiempo, la relación virus-hospedero pase del parasitismo (el virus se beneficia dañando al hospedero) al comensalismo (el virus se beneficia sin dañar al hospedero) e incluso al mutualismo (virus y hospedero se benefician mutuamente).
¿Qué provoca que un virus pase de animales a humanos?
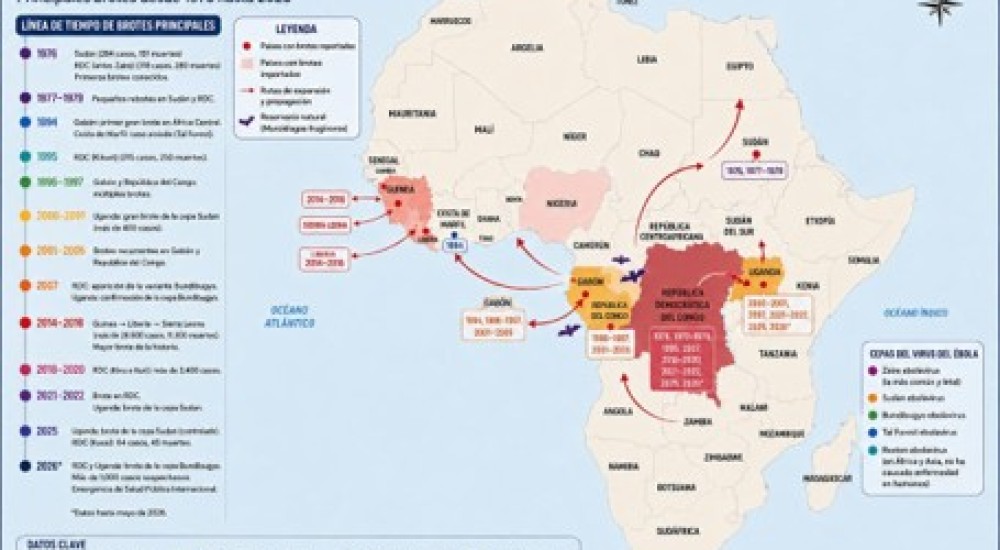
Hay varios factores que influyen sobre la probabilidad de una zoonosis. Uno de ellos es la cercanía evolutiva entre el hospedero original y el nuevo hospedero. Por ejemplo, es más probable que el humano adquiera virus de chimpancé que un virus de abeja. Otro factor muy importante son las actividades humanas que propician la cercanía física entre las especies animales. La migración de especies silvestres que establecen un mayor contacto con el humano debido a la deforestación de los bosques tropicales, el mantenimiento de grandes poblaciones de animales de consumo humano con poca diversidad genética en espacios reducidos y con poca higiene, y el tráfico de animales silvestres, algunos en peligro de extinción, nos ponen en contacto con especies que podrían ser reservorios o intermediarios en el proceso zoonótico. Este podría ser el caso del nuevo coronavirus que pudo haber evolucionado en murciélagos y posteriormente transferido al humano a través de otro animal que actuó como especie intermedia. No obstante, el contacto cercano con un reservorio o la exposición frecuente al virus no garantiza el surgimiento de una relación estrecha entre el virus y su nuevo hospedero (Figura 1). El establecimiento exitoso de una infección posterior a la exposición depende de la infectividad del virus. Esto es, la capacidad del virus de invadir a un organismo. Para que esto suceda el virus se debe asociar a una célula susceptible (célula con receptores que se unen al virus) y permisiva (célula con la maquinaria necesaria para reproducir al virus). Luego, el virus tiene que ser capaz de transmitirse de manera eficiente entre individuos de la misma especie. Esto depende de su transmisibilidad (capacidad de pasar de un individuo a otro). Finalmente, el surgimiento de una epidemia y eventual pandemia depende tanto de las propiedades de la población hospedera, como su tamaño y densidad, como de las propiedades biológicas del virus, como la virulencia (capacidad de producir una enfermedad). Si un virus es muy letal, no tendrá mucho éxito en poblaciones pequeñas y aisladas ya que si matan a todos los individuos de la población, no logrará propagarse hacia otras poblaciones. Por eso, se piensa que algunas enfermedades virales no se establecieron exitosamente en las poblaciones humanas hasta el origen de las civilizaciones. Con la aglomeración de las personas, la domesticación de los animales y el desarrollo del comercio que conectó a las poblaciones como nunca antes, se generó un ambiente propicio para la transmisión y propagación de patógenos como los virus. También, hay algunas enfermedades virales que facilitan la transmisión del virus como la tos seca, que es uno de los síntomas de la enfermedad por coronavirus de 2019 (COVID-19).
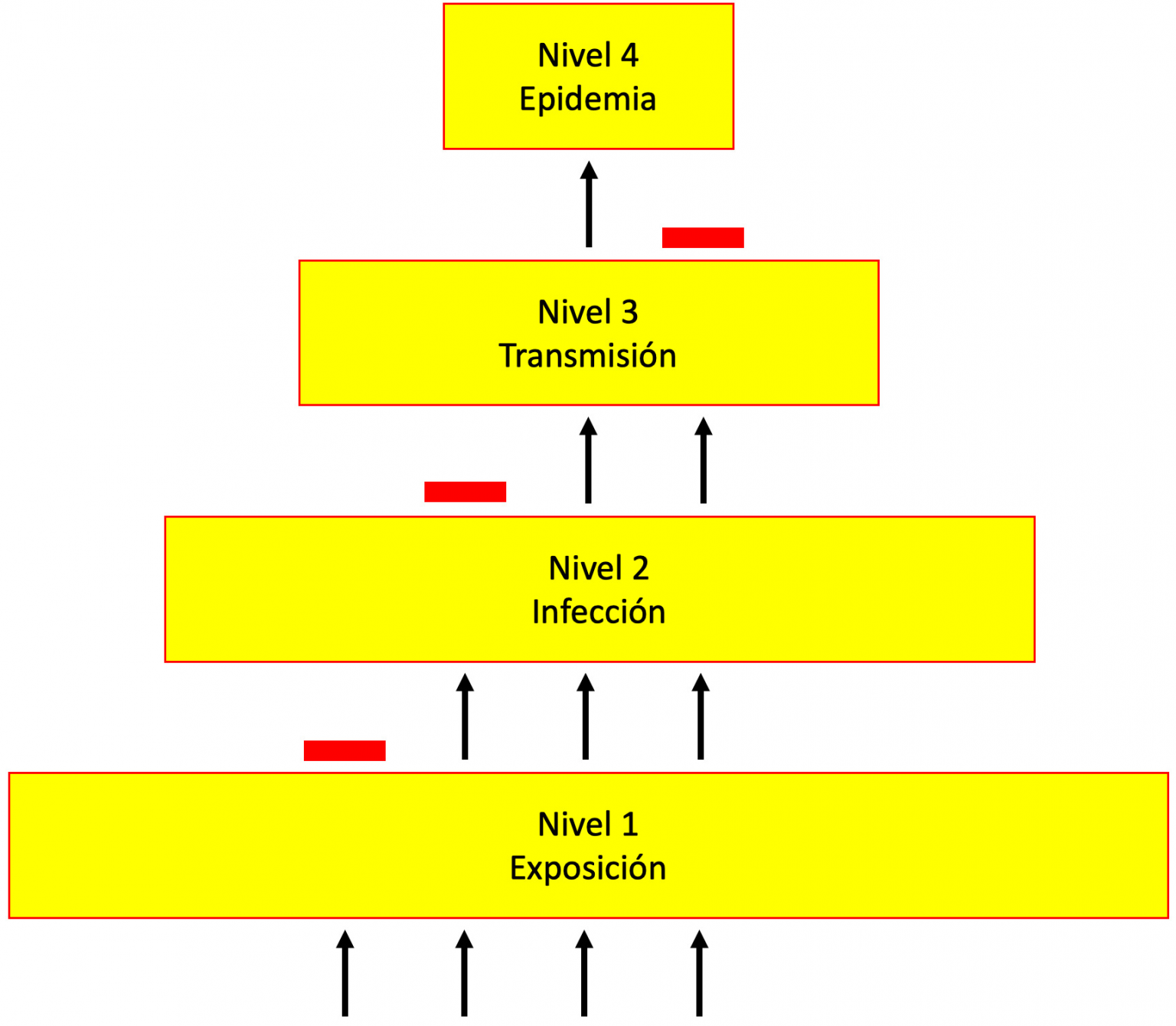
Figura 1. Niveles de interacción entre virus y hospedero. Algunos virus (flechas) alcanzan el nivel cuatro y otros son detenidos en diferentes niveles por barreras biológicas (barras rojas). Modificada de Woolhouse et al. (2012).
¿Cómo se pudo originar el nuevo coronavirus? Mitos y realidades del SARS-CoV-2
La comprensión del origen natural del nuevo coronavirus (SARS-CoV-2) se ha visto seriamente nublada por acusaciones derivadas de la tensión política y económica entre Estados Unidos y China. Esto se ve reflejado en funcionarios de ambos países responsabilizando, sin base alguna, al país adversario sobre el surgimiento del virus. De las diferentes explicaciones conspirativas hay dos que son defendidas y popularizadas por grandes grupos de fanáticos. La primera, que el virus fue diseñado y liberado de manera deliberada como un acto de bioterrorismo y, la segunda, que el virus fue creado en el Instituto de Virología de Wuhan de donde, por accidente, se escapó. Sin embargo, no hay evidencias que soporten estas explicaciones y la lógica detrás de ellas es muy debatible.
Por un lado, si el virus hubiese sido diseñado con fines terroristas o de guerra biológica se esperaría que se hubieran basado en la biología de algún virus ya conocido. Este virus debía tener capacidad de producir infecciones masivas en el humano, como el SARS-CoV, causante de la pandemia de 2002. Pero si analizamos el virus a nivel de su información genética, observamos pequeñas diferencias cuyo efecto hubiera sido imposible predecir. Si, por otro lado, el virus fuera el producto de una bienintencionada pero fallida investigación científica, de donde logró escapar a pesar de las medidas de bioseguridad, la comunidad científica conocería con detalle el origen directo del nuevo coronavirus. Ambas posibilidades se pueden desestimar si indagamos en el origen del virus. Esto se logra a través de los estudios genómicos (descrito en: http://www.acmor.org/articulo/la-situacion-actual-del-covid-19-en-mexico-y-en-el-mundo). En el caso del SARS-CoV-2 no se conoce un virus lo suficientemente parecido, y el SARS-CoV de 2002 presenta diferencias suficientes para descartarlo como base para el diseño de un nuevo virus. El virus más parecido al SARS-CoV-2 es un coronavirus de murciélago (RaTG13) que se le parece en un 96% (Figura 2) del cual se calcula que divergió hace unos 52 años. Si extrapolamos esta comparación con genomas de vertebrados, las diferencias equivaldrían a las de un cerdo y un humano, que están lejos de ser especies hermanas como el humano y el chimpancé. Este virus de murciélago se estudia en el Instituto de Virología de Wuhan, pero su relativamente lejano parentesco con el nuevo coronavirus de humano, indica que el virus que dio origen al SARS-CoV-2 es otro que sigue perteneciendo al amplio universo de virus desconocidos por la comunidad científica. Recientemente se descubrió otro coronavirus de murciélago (RmYN02) cercanamente relacionado con el SARS-CoV-2 con el que comparte el mayor porcentaje de identidad de la región 1ab que abarca más del 60% del genoma. Independientemente de las teorías de conspiración, la información filogenética reafirma que los murciélagos del género Rhinolophus son importantes reservorios de coronavirus y que son potenciales fuentes de zoonosis.
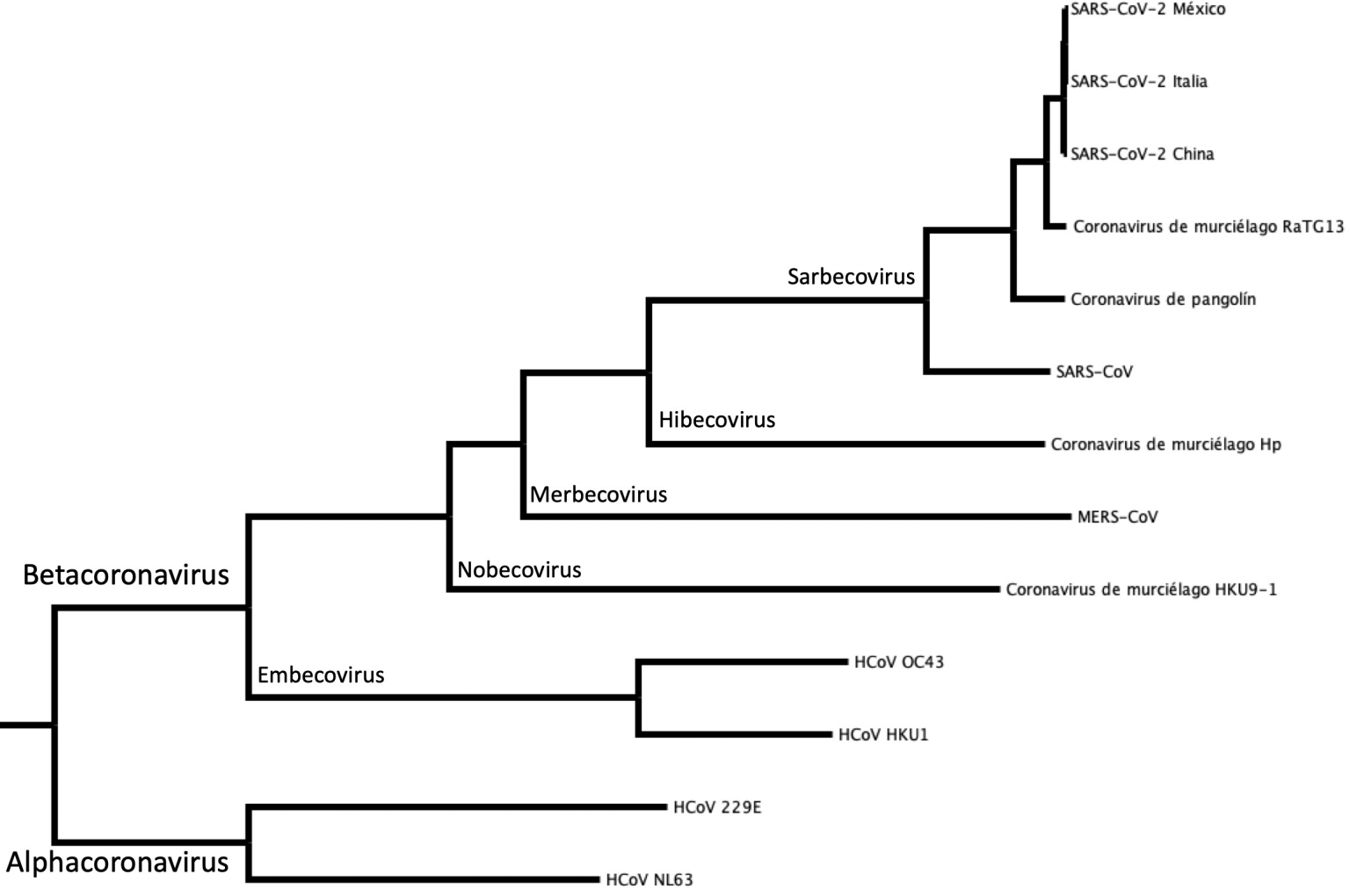
Figura 2. Relación evolutiva entre los coronavirus del género Betacoronavirus. Árbol filogenético de máxima verosimilitud realizado con los genomas completos de 13 coronavirus. Cada par de ramas parte de un nodo (origen) común. Cada nodo representa a un ancestro común. La distancia al nodo refleja el tiempo de divergencia desde el ancestro. Por ejemplo, SARS-CoV-2 de México tiene un ancestro común más cercano con el SARS-CoV-2 de Italia que con el MERS-CoV. Significa que ha pasado menos tiempo desde que las variantes de SARS-CoV-2 de México e Italia se separaron a partir de una misma población.
Las evidencias a nivel molecular de la evolución del virus
Los coronavirus tienen una proteína que sobresale alrededor de la partícula viral que, visto al microscopio, le da la apariencia de una corona solar. A esta proteína se le conoce como espícula o S (por spike en inglés). La espícula está compuesta de dos subunidades: la S1, que se une a las células; y la S2, que al ser cortada por una proteína celular permite que el genoma del virus entre a la célula (Figura 3) (ver más detalles en: http://www.acmor.org/articulo/covid-19-avances-y-perspectivas). En el SARS-CoV-2 se han observado características particulares asociadas a ambas subunidades:
En la subunidad S1, hay una región conocida como dominio de unión al receptor o RBD, por sus siglas en inglés. El RBD de SARS-CoV-2 tiene una combinación de aminoácidos diferente a la del SARS-CoV. Se sabe que la combinación de aminoácidos del SARS-CoV le permite unirse a las células humanas con una buena eficiencia. En un experimento en el que se estaba estudiando la eficiencia, se probaron diferentes combinaciones en la subunidad S1 del coronavirus de civeta (una especie de carnívoro distribuido en el sudeste asiático). Eventualmente se encontró la combinación más eficiente para unirse a las células de humano. Si el nuevo coronavirus fuera una creación de laboratorio, lo más seguro es que nuestros diseñadores hipotéticos hubiesen utilizado la combinación óptima. La combinación de aminoácidos del SARS-CoV-2 es más eficiente que la del SARS-CoV, pero está lejos de ser la óptima obtenida en los experimentos. Adicionalmente, la combinación de aminoácidos observada en el SARS-CoV-2 es parecida a la observada en el coronavirus de pangolín, lo cual nos indica que este tipo de mutaciones en la S1 pueden surgir de manera natural.
En la interfaz entre S1 y S2 hay cuatro aminoácidos adicionales que no se encuentran en los coronavirus más parecidos. Esta secuencia particular de aminoácidos es reconocida por otra proteína celular que genera un corte que separa a ambas subunidades (el posible rol biológico de esta inserción se puede ver en: http://www.acmor.org/articulo/covid-19-seis-meses-del-inicio-de-la-pandemia-primera-parte). En experimentos de laboratorio con el virus de la influenza, se ha observado que este tipo de inserción incrementa la patogenicidad del virus. Sin embargo, esta característica también ha surgido de manera natural en coronavirus de un subgénero evolutivamente más distante (Embecovirus) y en la misma región se ha detectado una inserción similar en el recientemente descubierto coronavirus RmYN02. Además, uno de los aminoácidos adicionales produce un cambio estructural en la proteína que podría facilitar una modificación capaz de conferirle al virus la habilidad de evadir al sistema inmune. El surgimiento de este mecanismo de evasión tiene más sentido bajo la luz de la evolución, en un contexto de adaptación a las presiones de selección natural ejercidas por el sistema inmune del hospedero.
El hecho de que la proteína S del SARS-CoV-2 comparte características con la proteína S del coronavirus de pangolín sugiere que pudo haber tomado elementos de otros virus por recombinación, un mecanismo natural de evolución que es frecuente en virus de RNA (ver: http://www.acmor.org/articulo/covid-19-seis-meses-del-inicio-de-la-pandemia-primera-parte). Sin embargo, un estudio reciente muestra evidencias de que el SARS-CoV-2 y los coronavirus de pangolín probablemente descienden de un linaje de coronavirus de murciélago, cuyo ancestro tenía un RBD con una combinación de aminoácidos que le permitía infectar a diferentes especies de mamíferos. Virus como el RaTG13 o el RmYN02 habrían adquirido otra combinación de aminoácidos en esta región posiblemente por recombinación con otros coronavirus aun desconocidos.
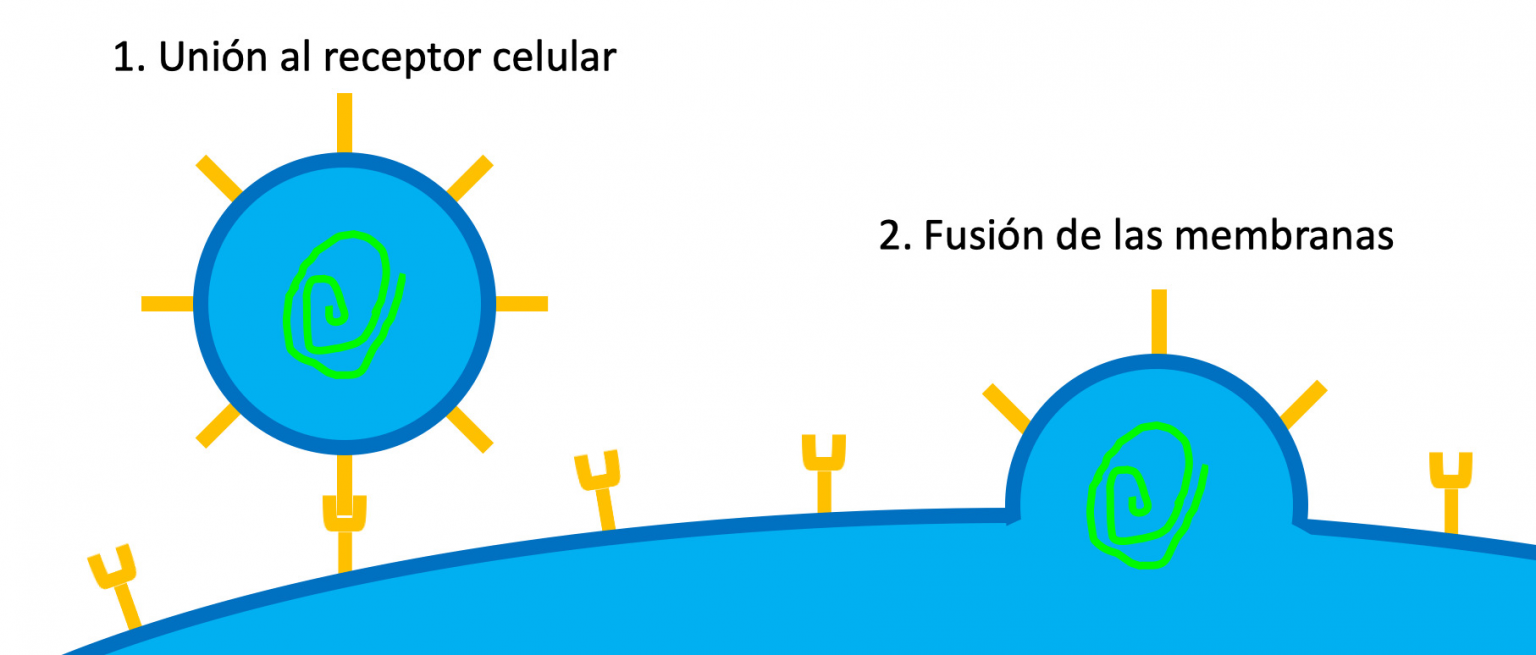
Figura 3. Caricatura del mecanismo de entrada del coronavirus a la célula. Para que el virus se una a la célula la subunidad S1 de la proteína S tiene que interactuar con un receptor celular. El procesamiento de la subunidad S2 induce la fusión de la membrana del virus con la membrana celular, lo que permite liberar el genoma viral en el citoplasma.
Conocer el origen de los virus no es una mera curiosidad. En el caso del nuevo coronavirus esto implica detectar tanto al hospedero original o reservorio como a la especie intermedia. Estos pudieron haber sido los murciélagos y los pangolines, respectivamente. No obstante, hoy en día no hay evidencias suficientes para probar esto, pero tener esta información nos permitirá entender las causas del surgimiento del nuevo virus y por lo tanto, desarrollar estrategias para evitar futuros brotes de virus similares.
¿Es cierto que el coronavirus está mutando?
Sí. De hecho, las mutaciones son lo que nos permite estudiar la evolución del virus y rastrear la procedencia de las diferentes variantes que llegan a cada país, y son un proceso natural esencial para la evolución de las especies. Todos los seres biológicos mutan y evolucionan, algunos más rápido que otros. Por ejemplo, dada la tasa de mutación del humano, se espera encontrar cerca de 3 cambios al año en un genoma de más de 3 mil millones de pares de bases. En cambio, si tuviéramos la tasa de mutación del SARS-CoV-2, tendríamos alrededor de 3 millones de cambios al año, esto es el 1% de nuestro genoma. Parece una cifra alarmante, pero hay que considerar que el genoma del nuevo coronavirus es mucho más pequeño que el del humano, pues tiene aproximadamente 30 mil pares de bases. Por lo tanto, dada su tasa de mutación, se espera que estos virus acumulen cerca de 30 cambios en un año, apenas el 0.1% de su genoma. La mayoría de las variantes que se han analizado hasta ahora tienen 10 o menos mutaciones acumuladas y muy pocas han alcanzado hasta 20. También es importante mencionar que hay virus de RNA con tasas de mutación que superan a la de los coronavirus por uno o dos órdenes de magnitud. Esto significa que, comparado con otros virus de RNA, el nuevo coronavirus evoluciona de manera lenta. Esta es una buena noticia, pues, a diferencia de lo que ocurre con la influenza, es posible que una vez desarrollada la vacuna para el SARS-CoV-2, se pueda generar una inmunidad que podría durar varios años o incluso, por el resto de nuestras vidas. Estudiar la evolución del virus también tiene aplicaciones prácticas. Por ejemplo, al hacer la comparación de las secuencias de las diferentes variedades de SARS-CoV-2 se pudo inferir que el primer caso confirmado en Estados Unidos provenía de China. Entonces, Estados Unidos tomó la decisión informada de cerrar los vuelos provenientes del país asiático. Luego de comparar más genomas de coronavirus en Estados Unidos, se dieron cuenta de que la mayoría de sus casos provenían de Europa, entonces decidieron limitar los vuelos con Europa. Como podemos notar, las mutaciones no son necesariamente un motivo de alarma. Y, si bien hay investigaciones recientes que sugieren el surgimiento de nuevas cepas con mayor infectividad o virulencia, lo cierto es que no se tienen evidencias contundentes que soporten dicha suposición. Algunas de estas sugerencias se han basado en solo una o dos mutaciones, lo cual no es suficiente para el surgimiento de una nueva cepa. Para que haya una nueva cepa, los cambios en el genoma se deben ver reflejados en nuevas propiedades biológicas del virus, cosa que no se ha observado. Por lo tanto, hasta ahora, se considera que solo existen diferentes variedades de la misma cepa.
Finalmente, con la información disponible del virus ha sido posible desarrollar vacunas y antivirales, pero sobre todo saber de su origen y evolución nos permite tomar decisiones informadas para seguir su propagación y en el mejor de los casos evitarla. Es importante no solo continuar la investigación para combatir la enfermedad causada por este virus, sino también prevenir nuevas pandemias producidas por otros patógenos virales.
Esta columna se prepara y edita semana con semana, en conjunto con investigadores morelenses convencidos del valor del conocimiento científico para el desarrollo social y económico de Morelos. Desde la Academia de Ciencias de Morelos externamos nuestra preocupación por el vacío que genera la extinción de la Secretaría de Innovación, Ciencia y Tecnología dentro del ecosistema de innovación estatal que se debilita sin la participación del Gobierno del Estado.
Referencias
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature Medicine, 26, 450-452. https://doi.org/10.1038/s41591-020-0820-9
Boni, M. F., Lemey, P., jiang, X., Lam, T. T-Y., Perry, B. W., Castoe, T. A., Rambaut, A., & Robertson, D. L. (2020). Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsable for the COVID-19 pandemic. Nature Microbiology. https://doi.org/10.1038/s41564-020-0771-4
Weiss, R. A. (2002). Virulence and pathogenesis. TRENDS in Microbiology, 10(7), 314-317. https://doi.org/10.1016/S0966-842X(02)02391-0
Woolhouse, M., Scott, F., Hudson, Z., Howey, R., & Chase-Topping, M. (2012). Human viruses: discovery and emergence. Phil. Trans. R. Soc. B., 367, 2864-2871. https://doi.org/10.1098/rstb.2011.0354
Zhou, H., Chen, X., Hu, T., Li, J., Song, H., Liu, Y., Wang, P., Liu, D., Yang, J., Holmes, E. C., Hughes, A. C., Bi, Y., & Shi, W. (2020) Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2Cleavage Site of the Spike Protein. Current Biology, 30, 2196-2203. https://doi.org/10.1016/j.cub.2020.05.023
Lecturas recomendadas
Ansede, M., Galocha, A. & Zafra, M. (2020). ccu cgg cgg gca. Las doce letras que cambiaron al mundo. El país. https://elpais.com/elpais/2020/05/09/ciencia/1589059080_203445.html
Briones, C. & Peretó, J. (2020). El origen del coronavirus SARS-CoV-2, a la luz de la evolución. The Conversation. https://theconversation.com/el-origen-del-coronavirus-sars-cov-2-a-la-luz-de-la-evolucion-136897
Corum, J. & Zimmer, C. (2020). Así muta y se propaga el coronavirus. The New York Times. https://www.nytimes.com/es/interactive/2020/04/30/science/coronavirus-mutacion.html
Más publicaciones